In nursing care of CF patients, it is important adjust your approach based on the individual patient. Caregivers and other family may or may not be involved in the care. It is important to establish trust with the patient, and family members if applicable, to determine who will be involved in the patient care team. CF is often diagnosed in infancy, however we know that many people with CF live well into adulthood. Some of these adult patients may be treated in a pediatric setting for continuity of care. It is important to address and interact with these patients based on their developmental age, not the care setting. Many CF patients endure frequent medical appointments and hospitalizations throughout their lives. These patients may be understandably fed up with the routine and their feelings can be manifested in their interactions with nursing staff. It is important to remember, however, that some CF patients do not experience hospitalization until late in childhood or even into adulthood. This experience can be very frightening for the patient and family members. It is important for nursing staff to assess and treat the physical and emotional manifestations of the disease. A few areas of system-specific assessment of CF patients are listed below.
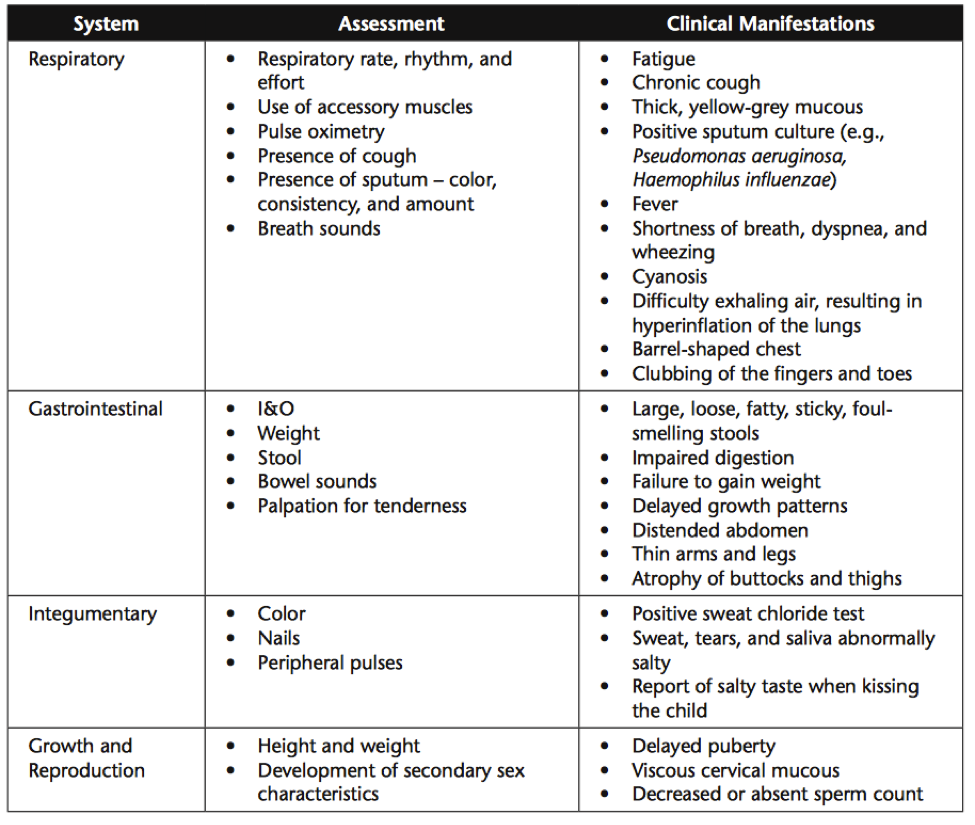
These assessments can require a wide variety of nursing interventions. Some of which include:
·
Respiratory
Interventions
·
Promptly
treat respiratory infections with antibiotic therapy.
·
Provide
pulmonary hygiene with chest physiotherapy (CPT) (e.g., breathing exercises to
strengthen thoracic muscles) a minimum of twice a day (in the morning and at
bedtime).
·
Have
the child use the Flutter mucus clearance device to assist with mucus removal.
·
Administer
bronchodilators through metered dose inhalers (MDIs) or hand-held nebulizer to
promote expectoration of excretions.
·
Administer
dornase alfa (Pulmozyme) through a nebulizer to decrease viscosity of mucus.
·
Promote
physical activity that the child enjoys to improve mental well-being,
self-esteem, and mucus secretion.
·
Gastrointestinal
Interventions
·
Administer
pancreatic enzymes with meals and snacks.
§ The amount of
enzyme replacement will vary between children based on each child’s deficiency
and response to the replacement.
§ Instruct the
child/family that the capsules can be swallowed whole or opened to sprinkle the
contents on a small amount of food.
§ Encourage the child
to select meals and snacks if appropriate.
§ Facilitate
high-caloric, high-protein intake through meals and snacks.
§ Multiple vitamins
and water-soluble forms of vitamins A, D, E, and K are often prescribed.
·
Hospitalization
·
The
child with cystic fibrosis is at an increased risk for hospitalization related
to pulmonary complications (e.g., respiratory infection, acute respiratory
distress).
·
The
child will receive respiratory treatments to include aerosol therapy, CPT,
breathing exercises, and assistance with coughing/expectoration of secretions.
·
Perform
CPT 1 hr before meals or 2 hr after meals if possible.
·
Use
oxygen with caution to prevent oxygen narcosis.
·
Promote
adequate nutritional intake, and provide pancreatic enzymes at meals and with
snacks.
·
Encourage
adequate fluid and salt intake.
·
Provide
meticulous skin care and oral hygiene.
·
Provide
encouragement and support to the child/family by using family-centered nursing
care.
·
Care
in the Home
·
Ensure
parents/caregivers have information regarding access to medical equipment.
·
Provide
teaching about equipment prior to discharge.
·
Instruct
parents/caregivers in ways to provide CPT and breathing exercises. For example,
a child can “stand on her head” by using a large, cushioned chair placed
against a wall.
·
Administer
antibiotics through a venous access port. Parents/caregivers need instruction
in administration techniques, side effects to observe for, and how to manage
difficulties with the venous access port.
·
Promote
regular primary care provider visits.
·
Ensure
up-to-date immunizations with the addition of initial influenza vaccine at 6
months of age and then a yearly booster.
·
Encourage
regular physical activity.
·
Encourage
participation in a support group(s) and involvement in community resources.
Overall, it is important to treat each patient as an individual, advocate for your patient, and equip the patient (and family if applicable) with the knowledge and support to maintain the best possible disease management at home.
Hockenberry, M., Wilson, D., Winkelstein, M. (2005). Wong’s essentials of pediatric nursing
care. (7th ed.). St. Louis, MO: Mosby.
http://www.atitesting.com/ati_next_gen/FocusedReview/data/datacontext/RM%20NCC%20PN%207.1%20Chp%2019.pdf




